
Thalassemia: What we need to know
DIVASHINI VIJIAN
UNIVERSITI SAINS MALAYSIA
divashinidiva@yahoo.com
Thalassemia is a common genetic disorder that affects 4.4 out of every ten thousand live births annually. Its severe type may cause transfusion-dependent anemia. Thalassemia is inherited in an autosomal recessive pattern; thus males and females are equally affected, as it does not involve the sex chromosome. The presence of two mutated genes, one inherited from each parent, allows for the trait to develop and become evident. Figure 1 shows genetic inheritance with carrier parents.
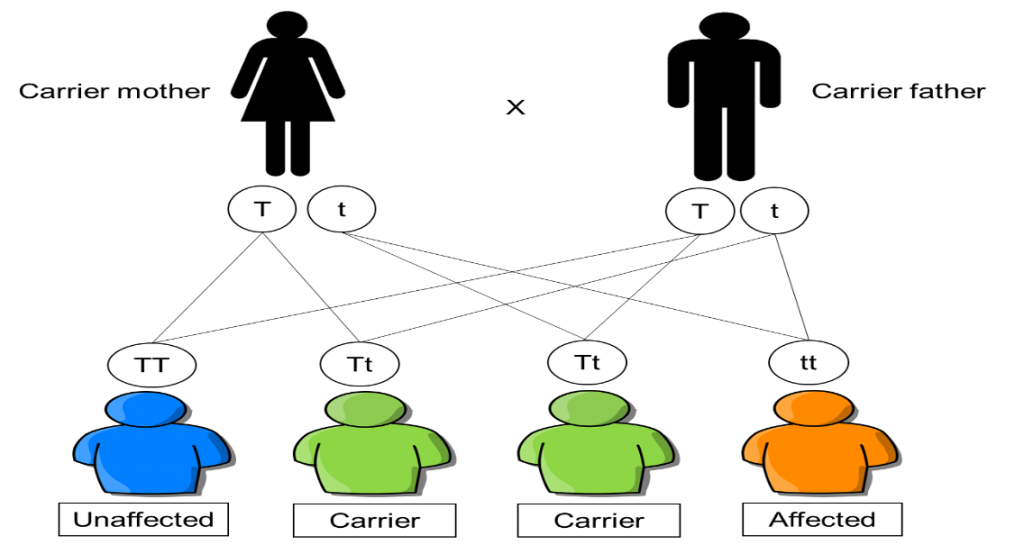 Figure 1: Genetic inheritance of Thalassemia
Figure 1: Genetic inheritance of Thalassemia
Thalassemia occurs when the body is unable to produce sufficient amounts of haemoglobin (Hb) due to a mutation in the genes coding for globin chains which lead to anemia. By definition, anemia is a condition reflected by the decrease in the red blood cells (RBCs) or Hb production in the body. It is closely associated with thalassemia, as it happens when there is an insufficient amount of alpha (α) or beta (β) globin chain production, an important component for making Hb. In healthy adults, the major components of Hb (Hb A) are two α and two β globin chains with heme (ring with iron) found in the RBCs. The structure of haemoglobin is shown in Figure 2. The primary function of RBCs is to transport oxygen from the lungs to all parts of the body. When there is a reduction in the amount of Hb there is an inadequate number of RBCs.
In normal circumstances, healthy individuals will inherit four functional α genes (αα/αα) and two functional β genes (β/β). However, in patients diagnosed with thalassemia, a genetic modification occurs leading to different types of thalassemia which are α, β, and delta (δ)-thalassemia or combination of these. However, the most common types are α and β-thalassemia. According to the World Health Organization, at least 20% of the world population carry mutated α gene, while 1.5% population globally carry mutated β gene. Alpha-thalassemia is prevalent in Africa, Southeast Asia, and the Middle East, while β-thalassemia is common in countries around the Mediterranean, the Middle East, parts of Central Asia, India, southern China, and the northern states. Alpha and β-thalassemia occurs due to the mutation in HBA and HBB genes located on chromosome 16 and 11, the mutations will cause a reduction in the synthesis of α and β globin chains, respectively.
The severity of this disease depends on the number of affected genes in an individual. Thus, clinical outcomes can be divided into thalassemia minor, intermedia, and major. Thalassemia minor patients usually will be asymptomatic or exhibit mild anaemia with no significant clinical outcome. Patients who are diagnosed with thalassemia major are often presented with more severe forms of clinical manifestations. Patients with thalassemia major will require regular blood transfusion as the body is unable to produce a sufficient amount of Hb. Blood transfusion provides blood components which are able to boost up the Hb level. Though blood transfusion cannot cure thalassemia permanently, yet, it can help in reducing the significant clinical signs and symptoms. In some cases, chelation therapy is required for the patients to remove excess iron in the blood that builds up as a result of regular blood transfusions.
The iron chelator commonly used were deferoxamine, deferiprone and deferasirox. The therapy of thalassemia focuses on the correction of the globin chain imbalance, improving iron overload and ineffective erythropoiesis. In this modern era, stem cell transplantation has been contemplated to treat this genetic disorder where the haematopoietic stem cells (HSC) are obtained from a healthy donor matching with the recipient. However, a major obstacle in this transplantation is the limited number of matched donors and the risky procedure involved. Gene therapy involves the autologous harvested from the patients, then the stem cells will undergo genetic modification with a lentiviral vector expressing a normal globin gene.
After a few proper therapies to destroy the existing defective HSC, the modified cells are introduced in the patients. Besides, thalassemia can also be treated by gene editing with few techniques such as zinc-finger nucleases (ZFN), transcription activator-like effectors nucleases (TALEN), or the clustered regularly interspaced short palindromic repeats (CRISPR) with Cas9 nuclease system. These techniques can replace the target mutated site with a corrected sequence. In current, pre-implantation genetic diagnosis (PGD) is conducted to determine if the embryo carries any gene abnormalities resulting in disease such as thalassemia. The early embryo is tested by removing one to two cells (blastomeres) at biopsy from the pre-implantation embryo at 6-10 cell stage, hence an embryo with abnormalities will be terminated from implantation into the uterus. The improvement in the understanding of thalassemia pathophysiology leads to various approaches to treat this genetic disorder.
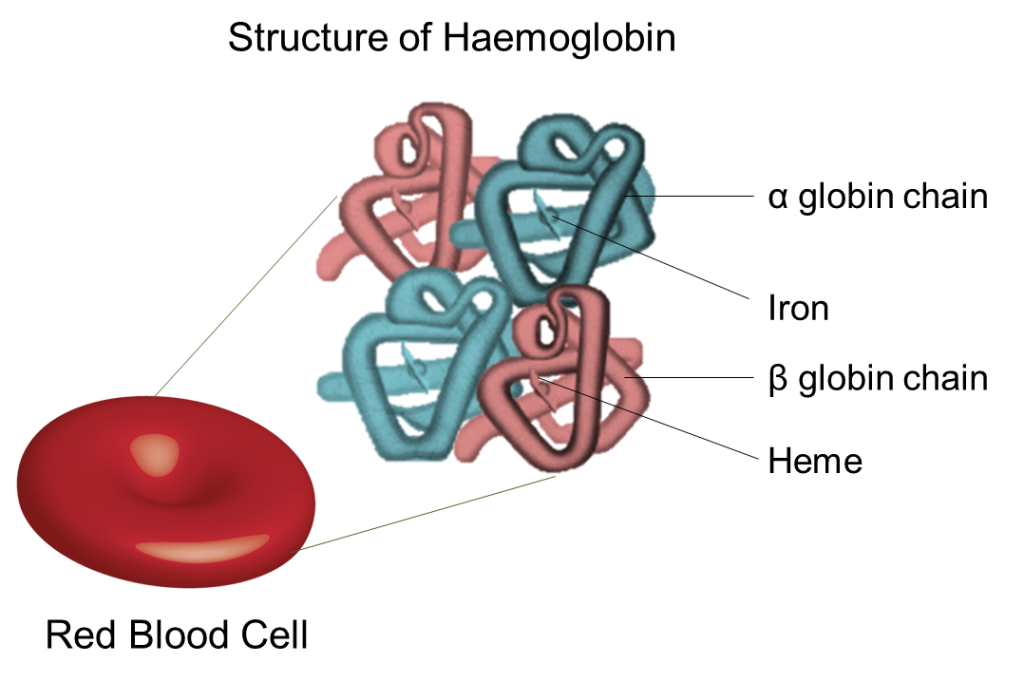
Figure 2: Haemoglobin structure comprising α and β globin chains
Reference (Apr-21-A8)
About the Author
Divashini Vijian is currently a postgraduate student at the School of Dental Sciences, Universiti Sains Malaysia. She obtained her Bachelor of Health Sciences (Honours) (Biomedicine) from Universiti Sains Malaysia. She has worked in the field related to haematology and specializes in the blood related genetic disorder, Thalassemia. Previously, she has worked on β-thalassemia to detect the common type of mutations detected in the studied community. Currently, she is working on the molecular and haematological characterization of α-thalassemia by using the latest molecular techniques to study the genetic sequence.
