
DIVERSITY OF lin GENES IN THE STRAINS ISOLATED FROM HCH CONTAMINATED SOIL
Researcher: Namrata Kundu, M. Tech Biotechnology, KIIT University
INTRODUCTION
Lindane also known as benzene hexachlorocyclohexane (BHC) is an organochlorine chemical
variant of hexachlorocyclohexane (HCH) that has been used as agricultural insecticide previously.
Technical HCH comprises four isomers (α, β, γ, δ) out of which only gamma (γ) isomer has
insecticidal property. Gamma isomer of HCH can be extracted and purified by photochlorination
of benzene under UV light which takes the form, commonly known to as lindane. Lindane enters
our food chain through agricultural water system. Lindane causes neurotoxin that interferes with
GABA neurotransmitter function by interacting with the GABAA receptor-chloride channel
complex at the ficrotoxin binding site. It affects the nervous system, liver and kidneys leading to
cancer hence, it is the need of the hour to treat HCH contaminants that enters our food chain and
causes diseases. Therefore, scientists have found a strain of bacteria Sphingobium indicum B90A
that has biodegradative and biosynthetic capabilities. Sphingomonas are gram negative bacteria,
rod shaped, aerobic contains glycosphingolipids (GSLS) instead of LPS and they produce yellow
pigmented colonies when grown in LB plates. Sphingomonas are subdivided into four genera:
Sphingobium, Novosphingobium, Sphingosinicella, and Sphingopyxis. This bacteria has got
immense application in bioremediation. They are also extensively used in food industries for the
production of extracellular polymers such as sphingans.
Among all other bacterial strains isolated from HCH contaminated soils, Sphingobium indicum
B90A appeared to have better potential for HCH degradation. This strain contains lin genes which
maybe scattered or organized in different operons. The transcriptional units that encode for the
lindane degradation pathway are lin A, lin B, lin C, lin DER, lin E. However, bioremediation for
decontamination of HCH by S. indicum B90A has got some disadvantages which we will discuss
later in this article.
METHODOLOGY
Techniques used to screen different strains of microbes from HCH dumpsite samples are-
• Plate pouring method
• Serial dilution and spread plate
• Streaking method
• Genomic DNA isolation by small scale supercos method
• Nanodrop spectrophotometer to find out the concentration and absorbance of DNA
• Gel electrophoresis method
• Colony PCR
PLATE POURING METHOD-
• We need to make LB media for 300 ml so weigh 6 grams LB in weighing balance.
• In a reagent bottle transfer the LB and weigh 4.5 grams Agar-Agar type I and transfer it
into the bottle.
• Measure 300 ml distilled water and transfer it into the bottle.
• Autoclave it.
• After cooling add nystatin (600 µl for 300 ml) and streptomycin (180µl for 300 ml).
• Pour the contents in the petri plates and keep it overnight.
SERIAL DILUTION AND SPREAD PLATE-
• Take six test tubes and label it.
• Weigh 0.9 gm NaCl.
• Measure 100 ml MQ/DW and put the weighed 0.9 gm Nacl into it in a 250ml bottle.
• Put 5 ml freshly prepared saline in each test tube.
• Put cotton plugs in each test tube and autoclave it.
• Now for the serial dilution take six MCT tubes inside the laminar and mark it.
• Take 900µl saline in every MCT.
• Weigh 1 gm HCH soil sample and 10 ml saline and take 100µl of this sample and transfer
it in the first marked MCT. Then again take 100µl sample from the first MCT and transfer
it into the second MCT.
• Take the agar plates and put one drop of the saline and soil sample mixture in it and do
spread plate.
• After doing spread plate put paraffin around the agar plate and incubate it in the room
temperature.
STREAKING-
• Label the plates at the bottom.
• Sterilize the inoculating loop in the bunsen burner by putting the loop into the flame until
it is red hot. Allow it to cool.
• Pick out pure colony from the previously streaked plates with the sterilized loop and lift
the petri plate at an angle of 60℃.
• Streak from side to side in parallel lines.
• First streaked line should be the continuation for the second streaking then third and fourth.
Last streaked line should not be in continuation with the first streaked line.
• After streaking is complete put paraffin around the plates.
• Incubate at 37℃
• After incubation for 24 hours we can observe growth along the streaked lines.
• We record the growth of individual colonies, morphology, colour.
GENOMIC DNA ISOLATION BY SMALL SCALE SUPERCOS METHOD-
• 5 ml culture pellet/ colonies from a fresh plate was taken.
• Prepare lysis buffer and adjacent and aliquot of 200µl of it in each eppendrof. Lysis buffer
consists of ( 5M Nacl, 1M Tris Hcl, 0.5M EDTA, and adjust the volume with MQ)
• Dissolve the pellet in lysis buffer and add 2µl of proteinase k (20mg/ml) to each.
• Add 10-20µl SDS (10%) and keep at 50℃ waterbath for 10 minutes (check lysis in regular
interval).
• Once lysis has taken place solution becomes viscous and slurry.
• Make up the volume to 1 ml by adding MQ.
• Add 1 ml phenol chloroform (equal volume), lower layer in the bottle should be taken
(phenol).
• Vortex nicely and till solution becomes white.
• Centrifuge 10,000 rpm for 15 minutes at 12℃.
• Take upper layer in fresh Eppendorf and add 1 ml chloroform.
• Vortex and centrifuge at 10,000 rpm/10 minutes at 12℃.
• Take the upper layer in fresh ependrof and add equal volume of absolute alcohol and mix
nicely (cut the tips) so that during transfer cell experience less friction.
• 20µl of 5M Nacl is added and invert mixed.
• Centrifuge at 10,000 rpm for 10 minutes at 12℃.
• Decant the supernatant and add 700µl of 70% ethanol.
• Centrifuge at 10,000 rpm for 10 minutes at 12℃ spool out the DNA.
• Decant supernatant and air dry the pellet.
• Dissolve in 500µl MQ and keep it at 37℃.
• Store at 4℃.
NANODROP SPECTRPHOTOMETER METHOD-
• First we open the software ND 1000 and go to blank setup where we add 2µl MQ to the
lower pedestal of the nanodrop instrument.
• Then we go to nucleic acids and add one drop of the DNA to be measured an then click on
the measure button.
• Then we get the absorbance and concentration of the DNA sample.
GEL ELECTROPHORESIS METHOD-
• Weigh 0.8 grams agarose in weighing machine and transfer it in gel bottle.
• Add 100 ml 0.5X TBE in the gel bottle.
• Heat it in the oven for nearly two minutes then with silicon gloves take it out from the
oven, keep it in room temperature for sometime then add ethidium bromide to it and mix
well.
• Then cast the gel in the gel apparatus and wait for 30 minutes to solidify.
• After 30 minutes remove the comb slowly from the apparatus and load the DNA samples
into the well using loading dye for visibility.
• Run the gel at 100V and after running it for few hours when the samples come out of the
wells then run it at 70V for proper resolving.
• Then observe it in gel documentation and analyse the result comparing it with DNA ladder
and take image of the gel.
COLONY PCR METHOD-
• Pick up 14 cell isolates collected from HCH dumpsite and one B90A cell as positive control
they are labelled as (MSM2, MSM3, MSM4, DS9, DS11, DS12, DS1, DS3, DS8, S1,
DS15, DS14, DS19, B90A ) from the streaked or restreaked plates.
• Suspend in 40µl MQ in MCT tube inside the laminar.
• Boil for 10 minutes, snap chill for 5 minutes on ice.
• Flash spin the MCT tubes.
• Take out 30µl of supernatant and add 70µl MQ in fresh MCT.
• Use this as DNA for PCR.
• Prepare the master mix for PCR which includes the following-DNA soup(15µl, taq
buffer(5µl), DMSO(1.5µl), taq polymerase(0.5µl), dNTPS(3µl), forward primer(1.5µl),
reverse primer(1.5µl) and MQ(22µl).
• Then set the programming in PCR machine and run it
RESULT
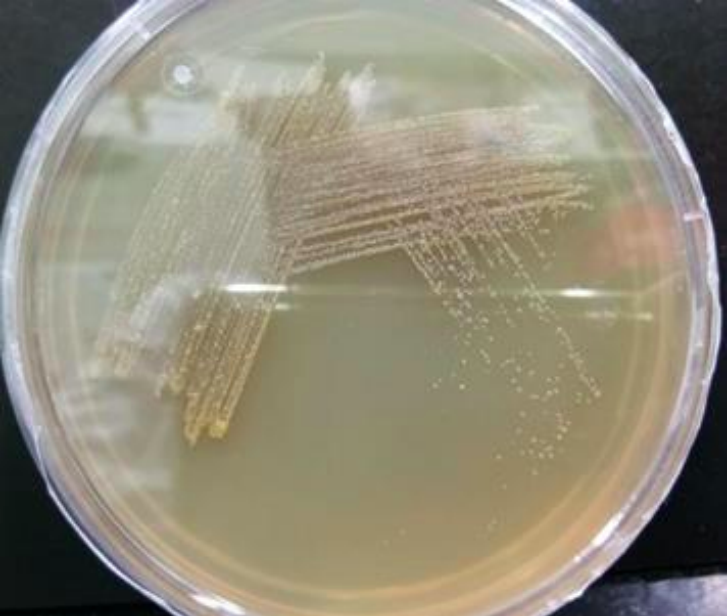
Streak plate- We observed brown color colonies, with slimy growth, this is a B90A strain.
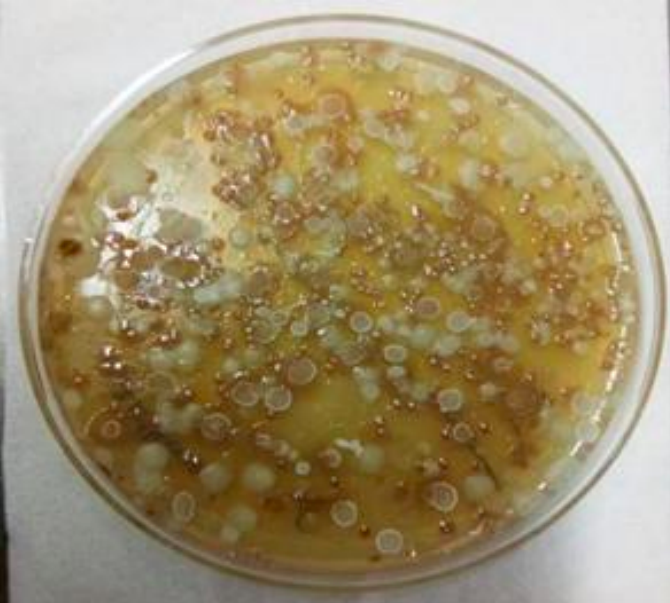
Spread plate technique- Colonies on agar plate after spread plate technique.
DISCUSSION
HCH degradation has become a very important issue for the environmentalist. Though in India
HCH usage is banned, still there are some parts where poisonous HCH is used as an agricultural
insecticide which enters our tropic chain. This issue has become a very big challenge to the
scientific community. Studies on HCH degradation by aerobic means have become a great success
but there are very few research and results found for anaerobic degradation. Also HCH isomer
complexity adds on to the problem. Sphingobium indicum B90A strain has got greater potential
for HCH degradation but this has also got some limitations when applied to large scale. The
significance of genetic variations in lin genes should be exploited more to gather more knowledge
on HCH degradation.
CONCLUSION
We screened different strains of microbes from HCH dumpsite but we came to the conclusion that
no lin genes were present in any of the strains. Therefore, in the strains that we selected do not
have biodegrading capability and thus we need to look for other strains.
Reference (Jul-20-A7)
